Die frühesten Darstellungen von Menschen mit Down-Syndrom finden sich auf Keramiken und in Form von Terracotta-Figuren aus der Zeit um 2500 vor Christus. In der Malerei sind Gesichtszüge des Down-Syndroms erstmals auf italienischen und niederländischen Gemälden des 15. und 16. Jahrhunderts zu erkennen.
Erste wissenschaftliche Beschreibungen der Symptome des Down-Syndroms erfolgten durch die französischen Ärzte und Psychiater Jean-Etienne Dominique Esquirol (1772-1840) und Edouard Séguin (1812-1880). Sie waren es auch, die erstmals in ihren Studien zwischen geistigen Behinderungen und psychiatrischen Erkrankungen unterschieden und spezielle Einrichtungen für geistig Behinderte gründeten.
Historische Einordnung und Namensgebung
Im Jahr 1866 veröffentlichte der englische Arzt John Langdon Down (1828-1896) eine sehr präzise Beschreibung von Symptomen und Charaktereigenschaften, die er bei einer Gruppe von mehr als 10 Prozent der von ihm betreuten geistig Behinderten beobachtet hatte. John Langdon Downs Söhne führten die Arbeit des Vaters fort; eines seiner Enkelkinder, das das Down-Syndrom hatte, lebte 65 Jahre und war ein guter Billard-Spieler.
John Langdon Down beschrieb die charakteristischen Gesichtsmerkmale als „Mongolian Type“, da er eine Ähnlichkeit zum Volksstamm der Mongolen sah. Dies prägte über Jahrzehnte die Bezeichnung des Down-Syndroms als „Mongolismus“ und unterstrich den zu dieser Zeit vorherrschenden Glauben an die unterschiedliche Wertigkeit der verschiedenen Ethnien. John Langdon Down selbst soll sich jedoch zu Lebzeiten von diesem Begriff distanziert haben.
Offiziell wurde die Bezeichnung „Down-Syndrom“ erst Anfang der 1960er Jahre eingeführt. Der Ausdruck „Mongolismus“ wurde zunehmend als diskriminierend und rassistisch wahrgenommen. Zudem hatte die Weltgesundheitsorganisation (WHO) einstimmig einen von Einwohnern der Mongolei gestellten Antrag angenommen, mit der Bitte, den Begriff „Mongolismus“ nicht mehr zu verwenden.
Frühe Förderung und medizinischer Fortschritt
Als Vorreiter der „modernen Behinderten-Pädagogik“ erkannte John Langdon Down die Bedeutung einer angemessenen Förderung von behinderten Menschen. In der von ihm gegründeten Einrichtung „Normansfield“ für geistig behinderte Menschen in Teddington gab es bereits eine große Aula für regelmäßige Theateraufführungen der Bewohner. Zudem dokumentierte Down Langzeitverläufe des Down-Syndroms und fotografierte Menschen mit Down-Syndrom in mehreren Lebensphasen (zum Beispiel „Mary“, siehe Fotos).

Durch den medizinischen Fortschritt, insbesondere die Operationsmöglichkeiten der angeborenen Herzfehler vieler Menschen mit Down-Syndrom, haben die Lebenserwartung und die Lebensqualität der Betroffenen deutlich zugenommen. Bei einer angemessenen Förderung von Beginn an steht Kindern mit Down-Syndrom häufig ein erfülltes Leben bevor. Dennoch brauchen viele während ihres gesamten Lebens individuell angepasste Hilfen.
Genetische Ursachen des Down-Syndroms
Im Jahr 1959 entdeckte der französische Genetiker Jérôme Lejeune (1926-1994), dass eine Überdosierung von genetischer Information des Chromosoms 21 zum Down-Syndrom führt. Er beschrieb die Trisomie 21 mit einem zusätzlich vorliegenden Chromosom 21. Eine solche sogenannte „freie“ Trisomie 21 findet sich bei etwa 95 Prozent der Menschen mit Down-Syndrom.
Chromosomenanomalien entstehen immer dann, wenn bei der Zellteilung entweder zu viel oder zu wenig Erbgut von den Eltern ans Kind weitergegeben wird. Beim Down-Syndrom, auch als Trisomie 21 bekannt, handelt es sich um einen genetischen Defekt, bei dem das Chromosom 21 nicht, wie eigentlich vorgesehen, nur zwei Mal, sondern stattdessen dreimal vorhanden ist. Dies zieht sowohl körperliche als auch kognitive Beeinträchtigungen für das Kind nach sich.
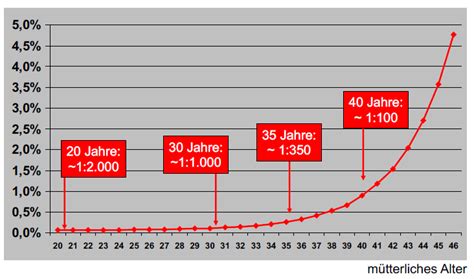
Die Rolle des mütterlichen Alters
Die Wahrscheinlichkeit, dass ein Kind mit einer freien Trisomie 21 geboren wird, steigt mit dem Alter der Mutter. Statistisch bekommt etwa eine von 100 Frauen im Alter von 40 Jahren ein Kind mit Down-Syndrom (1 Prozent). Dieses „Altersrisiko“ ist durch die zunehmende Anfälligkeit der Chromosomen für Fehlverteilungen in der Keimzellbildung bedingt. Mit zunehmendem Alter der werdenden Mutter steigt nun auch das Risiko einer Chromosomenanomalie wie dem Down-Syndrom. Je älter die werdende Mutter, desto höher die Wahrscheinlichkeit für Trisomie 21 beim Baby. So liegt beispielsweise die Wahrscheinlichkeit, dass eine 20-24-jährige Frau ein Baby mit Down-Syndrom zur Welt bringt, bei 1:1400, während die Wahrscheinlichkeit bei einer 45-Jährigen bei 1:25 ist.
Der Grund dafür ist der altersbedingte Abbau von Bindeproteinen, welche an Kraft verlieren oder abgebaut werden. Dies hängt unmittelbar mit dem Klebeprotein Cohesin zusammen, das sich in den Eizellen der Frau befindet und die Funktion hat, die in den Eizellen enthaltenen Chromosomen in Position zu halten. Es stabilisiert die Chromosomen auf diese Weise und sorgt für den richtigen Ablauf und das richtige Timing bei der Zellteilung. Mit zunehmendem Alter verliert das Klebeprotein Cohesin jedoch an Bindungskraft oder wird sogar gänzlich abgebaut. Das ist dadurch begründet, dass im Alter bestimmte Substanzen im Organismus schwinden, die das Cohesin vor Schäden und einem Abbau schützen. Das Altern und der damit einhergehende Zerfall des Cohesins können daher auch gleichzeitig das Auseinanderfallen der Chromosomen und die fehlerhafte Verteilung des Erbguts bei der Befruchtung bedeuten.
Andere genetische Ursachen
In selteneren Fällen (ca. 5 Prozent) wird das Down-Syndrom durch andere Chromosomenveränderungen von Chromosom 21 verursacht. Dazu zählen beispielsweise eine Translokations-Trisomie 21, Mosaik-Trisomie 21 oder ein Isochromosom 21. In diesen Fällen kann das Wiederholungsrisiko für weitere Kinder deutlich erhöht sein.
Diagnostische Verfahren
Nur eine Chromosomenanalyse bei einem Kind oder Erwachsenen mit Down-Syndrom bringt Klarheit, welche Form vorliegt und ob ein Vererbungsrisiko besteht. Diese Analyse unterscheidet zwischen erblichen und nicht-erblichen Formen des Down-Syndroms.
Pränataldiagnostik
Mit der Fruchtwasserpunktion und anschließender Chromosomenanalyse, erstmals durchgeführt 1966, wurde es möglich, eine Trisomie 21 und andere Chromosomenstörungen des Embryos sicher zu erkennen. Eine Fruchtwasserpunktion ist ab der 15./16. Schwangerschaftswoche möglich, und bis das endgültige Ergebnis vorliegt, vergehen meist zwei Wochen. Vorab kann ein „Schnelltest“ zum Nachweis bzw. Ausschluss von Trisomien der Chromosomen 13, 18 und 21 sowie zahlenmäßiger Veränderungen der Geschlechtschromosomen durchgeführt werden; das Ergebnis liegt nach einem Tag vor.
Alternativ kann bereits ab der 11. Schwangerschaftswoche eine Gewebeentnahme aus dem kindlichen Mutterkuchenanteil (Chorionzottenbiopsie) erfolgen. Eine Darstellung der kindlichen Chromosomen ist bereits nach ein bis zwei Tagen in der Kurzkultur möglich. Somit können Trisomien und Abweichungen der Geschlechtschromosomen schnell und sicher erkannt bzw. ausgeschlossen werden. In der Langzeitkultur, die meist nach zwei Wochen auswertbar ist, können zudem Strukturveränderungen der Chromosomen (z. B. Translokationen) erkannt werden.
Die Fruchtwasserpunktion und die Chorionzottenbiopsie sind invasive Verfahren, die mit einem geringen eingriffsbedingten Risiko für Komplikationen und Fehlgeburten einhergehen. Das Risiko, ein Kind infolge einer solchen Untersuchung durch eine Fehlgeburt zu verlieren, wird mit bis zu 0,5 Prozent (1:200) angegeben.
Werden eine Trisomie 21 oder andere Chromosomenstörungen festgestellt, ist grundsätzlich nach ausführlicher Beratung und Bedenkzeit ein Schwangerschaftsabbruch möglich, wenn die Schwangere dies wünscht.
Erst-Trimester-Screening
Das Erst-Trimester-Screening (ab 12. Woche) erlaubt eine Abschätzung, ob beim Embryo ein erhöhtes Risiko für eine Trisomie 21 vorliegt. Es ergibt sich ein individuelles Risiko für die jeweilige Schwangerschaft, das deutlich präziser als das alleinige altersbedingte Risiko ausfällt. Neben der Trisomie 21 (Down-Syndrom) wird auch ein Risikowert für die Trisomie 18 (Edwards-Syndrom) und die Trisomie 13 (Pätau-Syndrom) angegeben.
Beim Erst-Trimester-Screening wird mittels Ultraschall im Bereich des Nackens nach vermehrter Wassereinlagerung gesucht (Nacken-Transparenzmessung; NT-Messung). Zusätzliche Blutparameter der Mutter (biochemisches Screening: freies ß-hCG und PAPP-A) erhöhen die Genauigkeit. Hundertprozentige Sicherheit gibt das Erst-Trimester-Screening nicht, da ausschließlich etwa 90 bis 95 Prozent der Schwangerschaften mit einer Trisomie 21 im Erst-Trimester-Screening auffällig werden. Die Wahrscheinlichkeitsangabe des Erst-Trimester-Screenings dient jedoch vielen werdenden Eltern als Entscheidungshilfe für oder gegen eine invasive Diagnostik (Fruchtwasserpunktion oder Chorionzottenbiopsie).
Nicht-invasive Pränataltests (NIPT)
NIPT ist die Abkürzung für nicht-invasive molekulargenetische Pränataltests. Für deren Durchführung ist ausschließlich eine Blutprobe der Mutter (zwei Röhrchen) notwendig, somit besteht kein Fehlgeburtsrisiko durch die Untersuchung. Untersucht wird auf die kindlichen Trisomien 13, 18 und 21. Das sind die drei Trisomien, die mit steigendem Alter der Mutter häufiger werden und mit der Geburt eines lebensfähigen Kindes einhergehen können. Weitere Trisomien (z. B. Trisomie 16) führen dagegen praktisch immer zu einer Fehlgeburt, meist in der Frühschwangerschaft.
Seit 2012 werden in Deutschland NIPT-Verfahren hauptsächlich von drei Firmen angeboten. Bei den US-amerikanischen Firmen übernehmen deutsche Labore den Versand der Blutproben. Die Kosten müssen bislang selbst getragen werden, es kann jedoch bei der Krankenkasse ein Antrag auf Kostenübernahme gestellt werden. Das Ergebnis liegt meist nach zwei Wochen vor.
In der beginnenden 10. Schwangerschaftswoche (ab diesem Zeitpunkt werden die NIPT-Verfahren angeboten) enthält das Blut der meisten Schwangeren zwischen 4 und 20 Prozent kindliche DNA (sog. zellfreie fetale DNA). Auswertbar sind die Tests ab 4 Prozent kindlichem DNA-Anteil. Die Erbsubstanz wird vervielfältigt, sequenziert (einzelne Bausteine werden gelesen) und bioinformatisch ausgewertet. Dabei wird analysiert, ob die Dosis der Chromosomen 13, 18 und 21 im Vergleich zu anderen Chromosomen erhöht ist und somit der Hinweis auf eine Trisomie besteht.
Die Sicherheit der Tests kann methodisch bedingt nie 100 Prozent erreichen! Für die Trisomie 21 wird eine Sensitivität von 99 Prozent (1 Prozent falsch negativ) und eine Spezifität von 99,5 Prozent (0,5 Prozent falsch positiv) von den Firmen angegeben. Die neuen NIPT-Verfahren bergen die Gefahr, als „Screening-Tests“ ohne nennenswerte Risiken bereits früh in der Schwangerschaft „unkritisch“ und „großflächig“ eingesetzt zu werden. Bisher ist der erwartete „breite Ansturm“ jedoch ausgeblieben. Ein Teil der Schwangeren entscheidet sich sogar bewusst gegen jegliche vorgeburtliche Untersuchung. Andere empfinden die neuen NIPT-Verfahren als segensreiche Möglichkeit, die häufigsten Chromosomenstörungen untersuchen zu können, ohne das Risiko einer Fehlgeburt durch ein invasives Verfahren eingehen zu müssen.
NIPT-Verfahren werden weiterentwickelt; demnächst erfassen sie auch einige Mikrodeletions-Syndrome (z. B. DiGeorge-Syndrom: klinische Merkmale sind Herzfehler und variable Entwicklungsstörung). Denkbar sind längerfristig auch eine Dosismessung über das gesamte Genom (gesamtes Erbmaterial/alle Chromosomen) und Einzel-Gen-Analysen. Werdende Eltern benötigen also immer mehr Informationen, um sich individuell entscheiden zu können.
Merkmale und Fakten des Down-Syndroms
Das Down-Syndrom ist eine häufige genetische Ursache einer geistigen Entwicklungsstörung (ca. 1 auf 700 Neugeborene).
Häufige Symptome und Beeinträchtigungen
- Äußere Merkmale: aufsteigende Lidachsen, kleine Hautfältchen im inneren Augenwinkel (Epikanthus), flaches Profil mit flacher hervorstehender Zunge, kleine weiße Bindegewebsknötchen auf der Iris (Brushfield-Flecken), kurzer Hals, kurze Finger und Zehen (Brachydaktylie), querverlaufende Furchen an den Handinnenflächen („Vierfingerfurchen“).
- Muskelschwäche (Muskelhypotonie): insbesondere bei Säuglingen und Kleinkindern.
- Herzfehler (ca. 50 Prozent): häufig sind Defekte der Herzscheidewand.
- Fehlbildungen des Magen-Darm-Trakts (ca. 10 Prozent): Fehlbildung des Zwölffingerdarms (Duodenalatresie) und fehlende Nervenversorgung im Dickdarm (Morbus Hirschsprung).
- Immunsystem: erhöhte Infektanfälligkeit bei vielen Kindern.
- Blutkrebs (Leukämie): bei etwa 1 Prozent.
- Unterfunktion der Schilddrüse (Hypothyreose): bei 30 Prozent der Erwachsenen.
- Erwachsenengröße: häufig zwischen 1,40 und 1,60 Meter.
- Lebenserwartung: durchschnittlich über 50 Jahre. Haupttodesursachen im Kindesalter sind schwere Herzfehler und Fehlbildungen des Magen-Darm-Trakts. Im Alter vermehrt Demenzerkrankungen (Morbus Alzheimer).

Andere Chromosomenanomalien
Neben der Trisomie 21 können auch andere Chromosomenanomalien zu Entwicklungsstörungen führen:
- Trisomie 18 (Edwards-Syndrom) und Trisomie 13 (Pätau-Syndrom): Bei diesen Erkrankungen liegen die Chromosomen 18 oder 13 dreifach vor. Die Kinder haben oft Fehlbildungen an Kopf und Gehirn, Herz, Gliedmaßen und/oder anderen Organen. Die meisten Kinder sterben bereits in der Schwangerschaft oder in den ersten Wochen nach der Geburt.
- Monosomie X (Ullrich-Turner-Syndrom): Diese Abweichung der Geschlechtschromosomen betrifft nur Mädchen. Die Mädchen haben entweder nur ein weibliches Geschlechtschromosom (X-Chromosom) oder eines der beiden X-Chromosomen ist verändert. Mädchen mit Turner-Syndrom sind kleiner als der Durchschnitt und fast immer unfruchtbar. Oft benötigen sie Medikamente, damit die Pubertät einsetzt. Die größten gesundheitlichen Einschränkungen sind Veränderungen am Herz und an den Blutgefäßen. Mädchen mit Turner-Syndrom haben eine durchschnittliche Intelligenz, aber häufiger Probleme mit abstraktem Denken und zum Beispiel eine Rechenschwäche.
- Klinefelter-Syndrom: Diese Störung ist eine Abweichung der Geschlechtschromosomen, die nur Jungen betrifft. Die Jungen haben zu viele Geschlechtschromosomen, meist ein zusätzliches X-Chromosom. Oft sind die Symptome so unauffällig, dass die Chromosomenabweichung unbemerkt bleibt. Die Jungen sind meist überdurchschnittlich groß. Die Hoden sind dagegen in der Regel klein und produzieren nicht ausreichend Testosteron, sodass die Betroffenen fast immer unfruchtbar sind. Jungen mit Klinefelter-Syndrom sind durchschnittlich intelligent, brauchen aber manchmal etwas mehr Unterstützung in ihrer motorischen und geistigen Entwicklung.
Häufigkeit anderer angeborener Fehlbildungen
In Europa gibt es ein zentrales Melderegister, das die Daten von Kindern mit angeborenen Entwicklungsstörungen sammelt und veröffentlicht. Am häufigsten werden dort Fehlbildungen des Herzens gemeldet (ungefähr 8 von 1000 Geburten). Am zweithäufigsten sind genetisch bedingte Erkrankungen bei etwa 6 von 1000 Geburten. Etwa 3 von 1000 Kindern kommen mit einer Fehlbildung der Nieren oder des Harnsystems auf die Welt und etwa 3 von 1000 Kindern haben eine Fehlbildung des zentralen Nervensystems.
Angeborene Herzfehler
Bei 80 von 10.000 schwangeren Frauen hat das Ungeborene einen Herzfehler, der sehr unterschiedlich ausgeprägt sein kann. Herzfehler wirken sich meist erst nach der Geburt aus, wenn sich der kindliche Kreislauf umstellt und das Neugeborene selbstständig atmen muss. Viele kindliche Herzfehler sind gut behandelbar, weshalb die Kinder oft ein weitgehend normales Leben führen. Manchmal können kindliche Herzrhythmusstörungen schon während der Schwangerschaft behandelt werden, indem die Frau Medikamente einnimmt. Außerdem kann es Sinn machen, das Kind in einer spezialisierten Klinik zu bekommen, wo es direkt nach der Geburt optimal unterstützt wird.
Fehlbildungen an Wirbelsäule, Rückenmark und Gehirn
Etwa 30 von 10.000 Neugeborenen haben eine Fehlbildung am zentralen Nervensystem. 5 von 10.000 Kindern haben einen offenen Rücken (Spina bifida), etwa ebenso viele haben einen sogenannten Wasserkopf (Hydrozephalus).
Wenn sich das Baby im Mutterleib entwickelt, sind Rückenmark und Gehirn zunächst nur als längliches, röhrenartiges Gebilde angelegt (Neuralrohr). Kommt es bei der Weiterentwicklung zu Fehlbildungen, werden sie deshalb auch als Neuralrohrdefekte bezeichnet. In der Folge kann es zum Beispiel zu einem offenen Rücken (Spina bifida) kommen. Je nachdem, wo sich der Defekt befindet, sind leichte Lähmungen möglich, die Probleme beim Gehen verursachen - bis hin zur Gehunfähigkeit. In der Regel haben die Kinder Entleerungsstörungen von Darm und Blase. Zusätzlich kann der Kopf stark vergrößert sein, wenn die Abflusswege des Hirnwassers gestört sind (Hydrozephalus bzw. Wasserkopf).
Solche Fehlbildungen werden oft früh erkannt, meist beim Ultraschall. Wichtig ist dann eine sorgfältige Geburtsplanung und eine kompetente medizinische Versorgung des Neugeborenen in einem Spezialzentrum. Inzwischen ist es möglich, den offenen Rücken schon vor der Geburt zu operieren. Die schwerwiegendste dieser Fehlbildungen ist die Anenzephalie. Dabei haben sich Teile des Schädelknochens und des Gehirns nicht entwickelt. Diese Kinder sterben kurz nach der Geburt.