Nach der Geburt sind Neugeborene durch mütterliche IgG-Antikörper geschützt, die vor der Geburt über die Plazenta übertragen wurden. Dieser passive Schutz hält etwa vier Monate an, danach übernimmt die eigene Antikörperproduktion des Kindes die Abwehr von Krankheitserregern. Gestillte Kinder erhalten zusätzlich durch IgA-Antikörper in der Muttermilch, insbesondere im Kolostrum, einen sogenannten „enterale Nestschutz“.
Grundlagen der Immunglobuline
Immunglobuline, auch Antikörper genannt, sind Glykoproteine, die von B-Lymphozyten und Plasmazellen produziert werden und für die humorale Immunität verantwortlich sind. Sie werden in fünf Klassen eingeteilt, die sich in ihrer Struktur, ihrem Molekulargewicht, ihrer Elektrophoresefraktion, ihrem Vorkommen, ihrer Serumkonzentration, ihrer Plazentagängigkeit, ihrer Komplementaktivierung und ihrer funktionellen Bedeutung unterscheiden.
| Merkmal | IgG | IgM | IgA | IgD | IgE |
|---|---|---|---|---|---|
| Molekulargewicht | 150000 Da | 970000 Da | 160000/385000 Da | 175000 Da | 190000 Da |
| Elektrophoresefraktion | γ1 / γ2 | γ1 | γ1- β | γ1 | γ1 |
| Vorkommen | Serum, Muttermilch | Serum | Serum | Serum | Serum |
| Normale Serumkonzentration (Erwachsene) | 700 - 1600 mg/dl | 40 - 230 mg/dl | 70 - 400 mg/dl | 4 mg/dl | 0,03 mg/dl |
| Plazentagängigkeit | Klassen-abhängig | nein | nein | nein | nein |
| Komplementaktivierung | Klassen-abhängig | ja | nein | nein | nein |
| Funktionelle Bedeutung | (protektive) Antikörper der sekundären Immunantwort | Primäre Immunantwort | Immunologische Schleimhautbarrieren | Antigen-induzierte Reifung von B-Zellen | Soforttypallergie, Immunabwehr von Parasiten |
Für die humorale Immunabwehr sind vor allem die Klassen IgG, IgA und IgM von Bedeutung. Klinisch relevant sind dabei überwiegend der IgG- und IgA-Mangel.
IgG und seine Subklassen
Das fetale IgG stammt aus dem mütterlichen Blut und erreicht etwa in der 26. Schwangerschaftswoche (SSW) die mütterliche Konzentration. Nach der Geburt wird das mütterliche IgG mit einer Halbwertzeit von etwa 30 Tagen abgebaut. Dieser Rückgang der Leihimmunität und die verzögerte Eigensynthese des Säuglings führen zu einer physiologischen „Mangelphase“, insbesondere im 3. bis 5. Lebensmonat.
IgG-Mangelsyndrome
IgG-Mangelsyndrome können angeboren oder erworben sein. Ein Beispiel ist der variable humorale Immundefekt (CVID), bei dem es zu einer deutlichen Verminderung mehrerer IgG-Subklassen kommt, oft kombiniert mit einem Mangel an IgA und IgM. Diese Hypogammaglobulinämie tritt häufig erst im 3. bis 4. Lebensjahrzehnt auf. Im Gegensatz zu seltenen angeborenen Agammaglobulinämien sind die B-Zellen beim CVID zwar in normaler Zahl vorhanden, produzieren aber weniger Antikörper.
Ein sekundärer Antikörpermangel kann durch chronischen Proteinverlust (z. B. bei Enteropathien oder nephrotischem Syndrom) entstehen, wobei alle IgG-Subklassen gleichermaßen betroffen sind. Auch bei chronischen Lymphadenosen (CLL) und Plasmozytomen kann ein sekundärer Immunglobulinmangel auftreten, wobei hier zunächst IgM und IgA, und zuletzt IgG betroffen sind.
Bedeutung der IgG-Subklassen
Die Untersuchung der IgG-Subklassen ist bei klinischem Verdacht auf ein Antikörpermangelsyndrom wichtig, da eine normale Gesamt-IgG-Konzentration einen Subklassenmangel nicht ausschließt. Die verschiedenen Subklassen haben unterschiedliche Funktionen:
- IgG1 und IgG3 sind maßgeblich an der T-Zell-abhängigen Immunantwort gegen virale oder bakterielle Proteine beteiligt. Ein Mangel kann zu schweren bakteriellen Infektionen führen.
- IgG2 ist entscheidend für die Reaktion auf Kohlenhydratantigene. Ein Mangel begünstigt Infektionen der Atemwege durch bekapselte Bakterien und kann mit Autoimmunerkrankungen einhergehen.
- IgG3 enthält Antikörper gegen bakterielle Proteine und virusneutralisierende Immunglobuline. Ein Mangel kann zu rezidivierenden Infektionen der oberen Atemwege, Asthma bronchiale und Durchfällen führen.
- IgG4 spielt eine Rolle bei Allergien, indem es spezifisches IgE auf Mastzellen blockiert. Ein isolierter Mangel ist oft ohne klinische Relevanz.
Pathologisch erhöhte IgG-Subklassen-Konzentrationen können bei chronischer Antigenstimulation auftreten, beispielsweise bei HIV-Patienten (erhöhte IgG1 und IgG3) oder bei allergischer Alveolitis (massiver Anstieg von IgG2).
| Subklasse | Serumkonzentration (%) | Serumkonzentration (mg/dl) | Komplementaktivierung | Plazentagängigkeit | Serumhalbwertszeit | Fc-Rezeptorbindung (Monozyten) | Fc-Rezeptorbindung (Mastzellen) | Fc-Rezeptorbindung (Neutrophile) | Fc-Rezeptorbindung (Lymphozyten) |
|---|---|---|---|---|---|---|---|---|---|
| IgG-gesamt | 100 % | 700 - 1600 | +++++ | +++ | 21-23 Tage | ja | nein | ja | ja |
| IgG1 | 60 - 70 % | 280 - 800 | ++ | +++ | 21-23 Tage | ja | nein | ja | ja |
| IgG2 | 14 - 20 % | 115 - 570 | + | ++ | 7-9 Tage | nein | nein | ja | ja |
| IgG3 | 4 - 8 % | 24 - 125 | +++ | +++ | 21-23 Tage | ja | nein | ja | ja |
| IgG4 | 4 - 6 % | 5,2 - 125 | nein | + | 21-23 Tage | nein | ja | nein | ja |
Bei Verdacht auf angeborene Immundefekte im Kleinkindalter wird die Vorstellung in einem spezialisierten pädiatrischen Zentrum empfohlen. Patienten mit rezidivierenden bakteriellen Atemwegsinfektionen, Sinusitis, Otitis media oder unklaren IgG-Mangelzuständen sollten untersucht werden. Bei gesichertem IgG-Subklassenmangel und Infektneigung kann eine IgG-Substitutionstherapie erwogen werden.
IgA und die Schleimhautimmunität
IgA ist essenziell für die Abwehr von Infektionen auf Schleimhäuten und in Körpersekreten. Ein kombinierter IgG- und IgA-Mangel ist bei der Hypogammaglobulinämie (CVID) häufig. Der selektive IgA-Mangel ist die häufigste genetisch bedingte Immundefizienz. Bei gestillten Kindern erfolgt ein verspäteter Anstieg, sodass die Diagnose eines IgA-Defektes erst nach dem 2. Lebensjahr gestellt werden kann.
Folgen eines IgA-Defekts
Ein IgA-Defekt kann zu chronisch rezidivierenden, teils schweren Infektionen der Atemwege führen. Auch Nahrungsmittelunverträglichkeiten und gastrointestinale Symptome, die einer Sprue ähneln, können auftreten. Häufig kommt es zu Lambliasis aufgrund einer abwehrgeschwächten Darmschleimhaut. Des Weiteren besteht eine Neigung zu Allergien.
Eine präventive Behandlung mittels IgA-Substitution ist beim IgA-Defekt nicht möglich, da der Anteil an IgA in Immunglobulinpräparaten gering ist und der vorwiegend im Serum vorkommende Subtyp das schleimhautassoziierte IgA nicht ersetzen kann. Patienten mit IgA-Defekt können Antikörper gegen IgA bilden, was bei Transfusionen zu heftigen allergischen Reaktionen führen kann. Bei notwendiger Immunglobulinsubstitution werden Präparate mit abgereichertem IgA vorsichtig subkutan appliziert. Die Therapie konzentriert sich meist auf eine frühe antibiotische Behandlung von Schleimhautinfektionen.
Die Plazentaschranke
Die Plazentaschranke, auch Blut-Plazenta-Barriere genannt, schützt den Fötus vor Metaboliten des mütterlichen Stoffwechsels und verhindert den Übertritt fetaler Proteine in das mütterliche Blut. Sie ist anatomisch so aufgebaut, dass sie selektiv permeabel für bestimmte Stoffe ist.
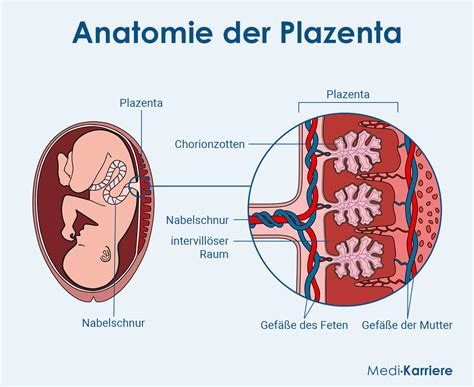
Stofftransport durch die Plazentaschranke
Gase, Wasser, Fettsäuren, Steroidhormone, lipophile Vitamine und Harnstoff werden durch Diffusion über die Plazentaschranke transportiert. Sauerstoffdiffundiert aufgrund der höheren Bindungsaffinität des fetalen Hämoglobins. Glukose wird über GLUT1-Transporter mittels erleichterter Diffusion geschleust. Calcium und Eisen werden aktiv transportiert, Proteine und LDL-Cholesterin über Endozytose.
Passiver Immunschutz durch IgG
Die Versorgung des Feten mit Immunglobulin G (IgG) erfolgt über die Plazenta durch Transzytose, die ausschließlich in maternofetaler Richtung stattfindet. Dies stattet das Ungeborene mit einer passiven Immunität gegen Krankheitserreger aus, gegen die die Mutter bereits Antikörper gebildet hat.
Physiologische und klinisch relevante Infektanfälligkeit
Eine physiologische Infektanfälligkeit ist Teil des normalen Reifungsprozesses des Immunsystems. Frühgeborene haben aufgrund der geringeren Antikörperübertragung einen weniger ausgeprägten „Nestschutz“. Mit zunehmendem Alter und der Entwicklung eines immunologischen Gedächtnisses nimmt die Infektanfälligkeit ab, meist ist dieser Zeitpunkt mit etwa dem 4. Geburtstag erreicht.
Diagnostik von Immundefekten
Bei Verdacht auf einen Immundefekt umfasst die immunologische Basisdiagnostik die Bestimmung des Blutbildes mit Differenzierung sowie von IgG, IgA und IgM. Dies kann bereits 60-70 % der Erkrankungen erfassen. Weiterführende Diagnostik in Spezialambulanzen umfasst die Bestimmung von IgE, IgG-Subklassen, Impfantikörpern und Komplementfaktoren.

Chronische Erkrankungen, insbesondere der Atmungsorgane, können einen Immundefekt vortäuschen oder Folge eines solchen sein. Bildgebende Verfahren und spezifische Tests (z. B. Schweißtest) sind hierbei zur Abgrenzung und Diagnostik notwendig.