Die Neurofibromatose Typ 1 (NF1), auch bekannt als Morbus Recklinghausen, ist eine genetisch bedingte Erkrankung, die vor allem die Haut und das Nervensystem betrifft und daher den neurokutanen Erkrankungen (Phakomatosen) zugeordnet wird.
Genetik und Vererbung
Eine Neurofibromatose wird durch eine Veränderung in einem Gen hervorgerufen, welches normalerweise hemmend auf die Zellteilung Einfluss nimmt. Dies führt zu überschießender Gewebsvermehrung und den charakteristischen Veränderungen. Etwa 50 Prozent der betroffenen Personen haben eine Neumutation, das heißt, die genetische Veränderung tritt spontan auf und ist nicht von den Eltern vererbt worden. Alle bisherigen Beobachtungen bestätigen den autosomal dominanten Erbgang, was bedeutet, dass ein betroffener Elternteil mit einer Wahrscheinlichkeit von 50 Prozent die Erkrankung an seine Kinder weitergibt. Es gibt keine unterschiedlichen Häufigkeiten in verschiedenen Regionen der Erde oder unter Angehörigen anderer ethnischer Gruppen, obwohl Männer etwas häufiger betroffen sind als Frauen.
Der Neurofibromatose-Typ-1-Lokus liegt auf dem Chromosom 17 Genlocus q11.2. Dieses Gen kodiert für das Protein Neurofibromin, welches eine wichtige Rolle bei der Regulation des Zellwachstums spielt. Neurofibromin zeigt Ähnlichkeiten mit dem GTPase-aktivierenden Protein (GAP) und stimuliert die GTPase-Aktivität von ras p21. Wenn GTPasen durch ihr GAP aktiviert werden, hydrolysieren sie GTP zu GDP, wodurch die Zellteilung stimulierenden Signale unterbrochen werden. Bei NF1 ist diese Regulation gestört, was zu unkontrolliertem Zellwachstum führt.
Klinisches Erscheinungsbild und Symptome
Die Diagnose wird meist anhand des klinischen Bildes bereits in der Kindheit gestellt. Zu den typischen Veränderungen an der Haut gehören Café-au-lait-Flecken und Neurofibrome.
Hautveränderungen
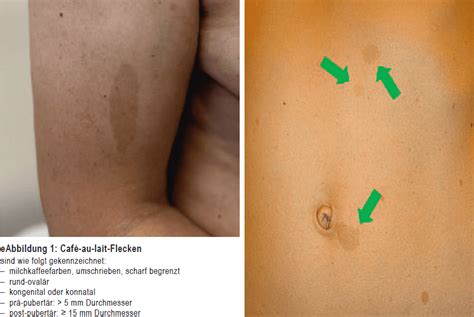
- Café-au-lait-Flecken: Dies sind milchkaffeefarbene Hyperpigmentierungen der Haut, die im Niveau der Haut liegen. Sie können bei allen Menschen auftreten und sind harmlos, treten aber bei NF1 gehäuft auf. In mehr als 95 Prozent der Fälle finden sich Café-au-lait-Flecken bei Patienten mit Neurofibromatose Recklinghausen. Etwa 80 Prozent weisen mehr als sechs große hyperpigmentierte Areale auf. Diese Flecken sind oft schon bei Geburt vorhanden und werden im Lauf der Kindheit häufiger und größer.
- Freckling: Eine sommersprossenähnliche Verfärbung an Körperstellen, die normalerweise keiner Sonnenbestrahlung ausgesetzt sind, insbesondere in der Achselhöhle und Leistenregion. Dies wird ab einem Alter von etwa 10 Jahren beobachtet und tritt bei etwa 90 Prozent der Patienten auf, was es zu einer diagnostisch wegweisenden Erscheinung macht.
- Lisch-Knötchen: Kleine, rundliche, scharf begrenzte und leicht erhabene Veränderungen in der Regenbogenhaut des Auges mit einem hellen, gelblich bis bräunlichen Farbton. Sie gelten als sehr hilfreiches diagnostisches Kriterium, da sie sich bei nahezu allen Patienten mit Neurofibromatose Typ 1 über 20 Jahren finden und ihre Anzahl mit dem Alter zunimmt.
- Neurofibrome: Gutartige Tumoren, die von den Zellen der Schwann’schen Scheiden kleiner Nervenfasern in der Haut ausgehen. Sie treten typischerweise kutan (in der Haut), subkutan (in der Unterhaut) oder als plexiforme Neurofibrome auf. Kutane Neurofibrome bilden sich typischerweise ab Beginn der Pubertät, plexiforme Neurofibrome sind oft angeboren. Die Haut kann im Lauf der Zeit mit bis zu 10.000 Tumoren unterschiedlicher Größe bedeckt sein. Sie variieren im Durchmesser von wenigen Millimetern bis zu mehreren Zentimetern und können unter der Oberfläche liegen oder halbkugelig aufsitzen. Ein charakteristisches Merkmal ist, dass sie bei Druck in die Tiefe ausweichen (Knopflochphänomen oder Klingelknopfphänomen).
Auswirkungen auf das zentrale Nervensystem und andere Organe
- Zentrales Nervensystem (ZNS): Im ZNS treten gehäuft Tumoren verschiedener Lokalisation auf. Patienten können aufgrund der Erkrankung geistig behindert (minderbegabt) sein und an epileptischen Anfällen leiden. Häufig sind auch Augen mitbetroffen, was zu Sehschwäche führen kann.
- Gliome: Den Großteil der bei NF1 auftretenden Gliome machen die im Bereich des Sehnervs (Nervus opticus) lokalisierten gutartigen pilozytischen Astrozytome aus, die auch als Optikusgliome bezeichnet werden. Diese treten charakteristischerweise bilateral auf und können über viele Jahre einen statischen Verlauf haben.
- Augen: Neben Optikusgliomen können Lisch-Knötchen auftreten.
- Knochen: NF1 kann schwerwiegende Auswirkungen auf die Knochenbildung haben. Dazu gehören die Dysplasie des Keilbeinflügels, die zu einer deformierten Augenhöhle führt, sowie Verformungen der langen Röhrenknochen der Extremitäten, die zu Brüchen neigen und Pseudogelenke bilden können. Im Kindesalter können sich Verformungen der Wirbelkörper zu einer schweren Skoliose entwickeln. Minderwuchs und eine Vergrößerung (Megalenzephalie) oder Asymmetrie des Kopfes sind ebenfalls belastende Symptome.
- Endokrines System: Die Pubertät kann verfrüht oder verspätet eintreten. Bei Hypothalamus-Hamartomen kann es zu einer Pubertas praecox kommen.
- Andere Tumoren: Das Auftreten von Phäochromozytomen (Tumoren des Nebennierenmarks) ist erhöht. Dasselbe gilt für andere seltene Tumoren wie Rhabdomyosarkome, juvenile Xanthogranulome, gastrointestinale Stromatumoren (GIST), medulläre Schilddrüsenkarzinome und chronische myelomonozytäre Leukämie.
Diagnose und Therapie
Die Diagnose wird meist anhand des klinischen Bildes gestellt. Als Kardinalsymptome oder Kernsymptome, durch deren gemeinsames Auftreten die Krankheit definiert ist, werden bei NF1 zwei Merkmale beschrieben. Zu den diagnostischen Kriterien zählen Symptome, die der allergrößte Teil der Patienten im Laufe der Erkrankung entwickelt. Dazu gehören Café-au-lait-Flecken und kutane Neurofibrome.
Da es sich bei Morbus Recklinghausen um eine genetische Erkrankung handelt, ist eine Therapie, welche ihre Ursache beseitigt, derzeit nicht möglich. Es werden daher nur Veränderungen behandelt, die für den Patienten störend oder gefährlich sind. Die einzige Behandlungsmöglichkeit besteht in der operativen Entfernung von Neurofibromen und Tumoren oder ausnahmsweise in deren Bestrahlung. Allerdings muss man hierbei sehr zurückhaltend vorgehen, da Operationen zu Funktionsausfällen und Lähmungen führen können und Bestrahlung ein vermehrtes Tumorwachstum auslösen kann. Tumoren des ZNS können so lokalisiert sein, dass eine operative Entfernung ohne Schädigung gesunden Gewebes nicht möglich ist. Daher ist eine genaue Risiko-Nutzen-Abwägung erforderlich. Üblicherweise werden nur Veränderungen entfernt, die ein Risiko einer bösartigen Entwicklung besitzen.
Epidemiologie und Lebenserwartung
Man schätzt etwa 30 bis 40 Erkrankte auf 100.000 Einwohner, was einer Erwartung von einem betroffenen Kind pro 2500 bis 3300 Geburten entspricht. Die Lebenserwartung bei Neurofibromatose Typ 1 ist im Allgemeinen nicht signifikant eingeschränkt, kann aber durch Komplikationen wie bösartige Tumoren oder schwere ZNS-Probleme beeinflusst werden. Statistiken zur Lebenserwartung sind schwierig zu erheben, da sie stark von der individuellen Ausprägung der Krankheit und dem Auftreten von Komplikationen abhängt.
Historische Einordnung
Eine eher anekdotische Erstbeschreibung findet sich bei Robert William Smith 1849. Friedrich Daniel von Recklinghausen legte 1882 die erste präzise klinische und pathologische Charakterisierung vor. Alex Thomsen gab um 1900 die ersten statistischen Daten und eine ausführliche Bibliographie heraus. Joseph Merrick, der sogenannte „Elefantenmensch“, galt lange Zeit als ein Beispiel für die entstellenden Auswirkungen der Recklinghausenschen Krankheit. Sein Leben im viktorianischen England war Grundlage für Bücher und Filme. Merricks schwere Entstellungen prägten die weitverbreiteten falschen Vorstellungen von der Monstrosität der Patienten mit einer Neurofibromatose Typ 1. Nach einer DNA-Analyse im Jahre 2003 litt Merrick aber am Proteus-Syndrom, wobei eine zusätzliche Erkrankung an Neurofibromatose Typ 1 wahrscheinlich ist.
tags: #neurofibromatose #lebenserwartung #neugeborenes