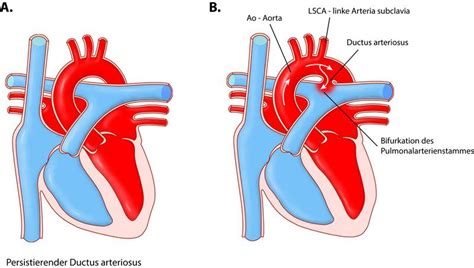
In der Gebärmutter verbindet ein spezielles Blutgefäß, der Ductus arteriosus, das Herz und die Lunge des Babys und sorgt für eine gute Durchblutung. Nach der Geburt verschließt sich dieses Gefäß normalerweise auf natürliche Weise, aber bei sehr frühgeborenen Babys kann es offenbleiben. Dies ist ein Zustand, der als persistierender Ductus arteriosus (PDA) bekannt ist.
Wenn der PDA klein ist, verursacht er möglicherweise keine Symptome. Ein größerer PDA kann jedoch zu einem erhöhten Blutfluss in die Lunge führen, so dass das Herz härter arbeiten muss. Dies kann zu Atemproblemen, Herzproblemen oder einer verminderten Blutzufuhr zu lebenswichtigen Organen wie den Nieren, dem Darm und dem Gehirn führen. Frühgeborene, insbesondere solche, die vor der 34. Woche geboren werden, haben ein 40%iges Risiko, diese Krankheit zu entwickeln. Unbehandelt schließen sich viele PDAs von selbst, aber das Behandlungsteam wird die möglichen Gesundheitsrisiken sorgfältig abwägen.
Die meisten Babys mit PDA haben ein Herzgeräusch, das bei Routineuntersuchungen festgestellt wird. Weitere Symptome können Atembeschwerden, abnormaler Blutdruck und Veränderungen der Herzfrequenz sein, die alle auf der Neugeborenenstation überwacht werden. Ein PDA kann sich bei richtiger Flüssigkeitszufuhr manchmal von selbst zurückbilden.
Herzfehler: Ein Überblick über angeborene Fehlbildungen
Herzfehler zählen zu den häufigsten angeborenen Fehlbildungen. Je nach Schweregrad der Fehlbildung und der damit verbundenen Symptome können einige Herzfehler ohne bedeutende körperliche Einschränkung jahrelang gut toleriert werden. Von großer Bedeutung ist die enge Zusammenarbeit der Bereiche Pränataldiagnostik, Geburtshilfe, Neonatologie, Kinderkardiologie und Kinderherzchirurgie. Somit kann eine möglichst frühe Diagnose erstellt werden, an die sich eine genaue Therapieplanung und operative Versorgung anschließt.
Bereits in den ersten Tagen der Schwangerschaft beginnt im Embryo die Entwicklung des Herz-Kreislaufsystems. In den nachfolgenden Wochen entwickeln sich durch komplexe Umbauvorgänge das Herz und die daran angrenzenden Gefäße. Betroffen sein können sowohl Strukturen innerhalb des Herzens, wie die Vorhof- oder Kammerscheidewand sowie die Herzklappen als auch die vom Herzen abgehenden Gefäße. Bleibt die Verbindung langfristig nach der Geburt geöffnet, besteht die Gefahr einer drohenden Lungenüberflutung mit Blut und einer daraus resultierenden Herzschwäche oder eines Lungenhochdrucks.
Spezifische Herzfehler und ihre operative Behandlung
Persistierender Ductus arteriosus (PDA)
Misslingt der Versuch einer medikamentösen Therapie, kann der Verschluss eines PDA über einen Herzkathetereingriff erfolgen.
Aortenisthmusstenose
Der Aortenisthmus ist der Bereich der Körperschlagader (Aorta) zwischen dem Beginn der linken Armarterie und der Einmündungsstelle des Ductus arteriosus. Es zeigt sich weiterhin eine oft schwere Herzschwäche. Neugeborene und junge Säuglinge müssen umgehend chirurgisch über einen Schnitt an der linken Brustkorbseite versorgt werden. Bei der Operation wird die Engstelle herausgeschnitten und die beiden Enden der Aorta durch eine Naht wieder miteinander verbunden.
Vorhofscheidewanddefekt (ASD)
Ein Vorhofscheidewanddefekt befindet sich zwischen dem rechten und linken Herzvorhof. Im kleinsten Fall handelt es sich um ein persistierendes Foramen ovale, wie es bei 30 Prozent aller Menschen, meist ohne Krankheitswert, vorkommt. Die Volumenüberlastung der rechten Kammer und des Lungengefäßbettes wird normalerweise lange ohne wesentliche Symptome toleriert.
Kammerscheidewanddefekt (VSD)
Ein Kammerscheidewanddefekt ist eine fehlerhafte Verbindung zwischen der linken und rechten Herzkammer. Der isolierte VSD ist mit bis zu 25 Prozent die häufigste Herzfehlbildung. Bei einer Volumenüberlastung entwickelt sich sehr schnell eine zunehmende Herzschwäche und ein Lungengefäßhochdruck.
Fallot'sche Tetralogie
Die Fallot'sche Tetralogie ist der häufigste Herzfehler, bei dem es zu einer Sauerstoffuntersättigung (Zyanose) kommt. Die Korrektur ist nur herzchirurgisch möglich. Über die Eröffnung des Brustkorbes werden Patient:innen an die Herz-Lungen-Maschine angeschlossen. Der Kammerscheidewanddefekt wird mit einem körpereigenen Herzbeutelflicken verschlossen, die überflüssige Muskulatur aus der rechten Pumpkammer wird abgetragen.
Atrioventrikulärer Septumdefekt (AVSD)
Bei einem Atrioventrikulären Septumdefekt kommt es zu einer Kombination von einem tiefen Vorhofscheidewanddefekt mit einem Kammerscheidewanddefekt. Es besteht eine gemeinsame Herzklappe zwischen den Herzvorhöfen und den Pumpkammern, die nicht selten eine Undichtigkeit zeigt. Die Korrektur ist nur herzchirurgisch möglich. Nach der Eröffnung des Brustkorbes werden Patient:innen an die Herz-Lungen-Maschine angeschlossen. Im Herzstillstand werden beide Defekte durch zwei Flicken getrennt voneinander verschlossen. Anschließend wird die gemeinsame Herzklappe in einen Mitral- und einen Trikuspidalklappenanteil getrennt.
Transposition der großen Arterien (TGA)
Bei einer Transposition der großen Arterien entspringt die Körperschlagader (Aorta) aus der rechten Herzkammer, während die Lungenschlagader (Pulmonalarterie) aus der linken Herzkammer entspringt. Das Überleben der Patient:innen ist nur möglich, wenn es zwischen den Vorhöfen, zwischen den Herzkammern oder über den Ductus arteriosus (PDA) Verbindungen zur Blutdurchmischung gibt. Durch eine meist noch im Neugeborenenalter erforderliche komplexe Operation am offenen Herzen unter Verwendung der Herz-Lungen-Maschine und im Herzstillstand werden die großen Körpergefäße und die Koronararterien herausgeschnitten und in die richtige Herzkammer implantiert. Danach befinden sich Lungen- und Körperkreislauf in physiologischer Reihenschaltung.
Unterbrochener Aortenbogen (IAA)
Bei einem unterbrochenen Aortenbogen liegt eine vollständige Unterbrechung zwischen verschiedenen Abschnitten des Aortenbogens und der absteigenden Aorta vor. Bei fast allen Patient:innen liegt außerdem ein Kammerscheidewanddefekt (VSD) vor. Unter Verwendung der Herz-Lungen-Maschine und im Herzstillstand wird der Aortenbogen rekonstruiert und eine Kontinuität geschaffen.
Truncus arteriosus communis (TAC)
Im Falle eines Truncus arteriosus communis entspringen die Körperschlagader (Aorta) und die Lungenschlagader (Pulmonalarterie) als ein gemeinsames Gefäß aus der Herzbasis. Die gemeinsame Herzklappe (Trunkusklappe) ist oft undicht und missgebildet und liegt in der Nähe eines Kammerscheidewanddefektes. Bei der chirurgischen Korrektur unter Verwendung der Herz-Lungen-Maschine werden im Herzstillstand die großen Lungengefäße aus der Aorta herausgeschnitten. Über eine biologische Gefäßprothese werden die Lungengefäße mit der rechten Pumpkammer verbunden.
Totale Lungenvenenfehleinmündung (TAPVR)
Bei einer totalen Lungenvenenfehleinmündung besteht keine direkte Verbindung zwischen den Lungenvenen, die sauerstoffreiches Blut aus der Lunge zum Herzen transportieren, und dem linken Vorhof. Bestehen in diesem Bereich Engstellen, kommt es nach der Geburt, ab der viel mehr Blut in die Lungen fließen muss als pränatal, zu einem kritischen Blutstau in der Lunge. In dieser Situation müssen die Patient:innen umgehend operiert werden.
Hypoplastisches Linksherz (HLHS) und Hypoplastisches Rechtsherz (HRHS)
Beim hypoplastischen Linksherz gibt es nur eine funktionierende (rechte) Herzkammer, die das Blut in die Lungenschlagader und über den Ductus arteriosus auch in die Körperschlagader pumpt. Bei dieser und ähnlichen sehr komplexen Fehlbildungen mit nur einer Herzkammer kann eine Korrektur nicht durchgeführt werden. Die chirurgische Behandlung von Herzen mit nur einer ausgebildeten Pumpkammer besteht in einer schrittweisen Trennung des kleinen und großen Blutkreislaufes. Die einzige vorhandene Herzkammer wird zur Durchblutung des Körperkreislaufs genutzt, der Lungenkreislauf wird durch Umgehung des Herzens passiv durchblutet. Das gesamte Konzept wird als „Fontan Zirkulation" bezeichnet.
Diagnostik und operative Versorgung
Die Befunde all unserer Patient:innen mit angeborenen Herzfehlern werden zur weiteren Therapieplanung wöchentlich in einer kinderkardiologisch-kinderherzchirurgischen Konferenz besprochen. Bei geplanten Operationen erfolgt die Aufnahme auf die kinderkardiologische Normalstation am Vortag der Operation. Sollten sich bei komplexeren Fehlbildungen dabei weitere Fragestellungen ergeben, können wir diese im Vorfeld des Eingriffs diagnostisch rund um die Uhr abklären, mit modernsten Verfahren wie Herzkatheteruntersuchung, Computertomographie und Magnetresonanztomographie.
Der Zugangsweg zum Herzen richtet sich nach der individuell vorliegenden Erkrankung. In den meisten Fällen erfolgt die Operation über eine mediane Sternotomie, das heißt, einen Zugang zum Herzen durch Eröffnung des Brustbeines, verbunden mit dem Einsatz der Herz-Lungen-Maschine. Am stillgelegten Herzen erfolgt die chirurgische Korrektur. Anschließend wird das Brustbein mit kleinen Drähten und die Haut mit dünnen Fäden sorgfältig verschlossen. Eltern und Angehörige informieren wir unmittelbar nach der Operation telefonisch über deren Verlauf.
Leben mit einem angeborenen Herzfehler
Jährlich kommen in Deutschland rund 8.000 Kinder mit einem Herzfehler zur Welt. Die Medizin rettet weit über 90 Prozent der kleinen Patienten heute das Leben. Darunter sind auch Menschen mit einem Einkammerherzen. Noch vor wenigen Jahrzehnten hatten Neugeborene mit solch einem komplexen Herzfehler keine Überlebenschance. Doch der Start ins Leben ist auch heute nicht leicht, die Perspektive zu Beginn oft ungewiss. Welche Krisen meistern diese Menschen - als Kinder, Jugendliche und Erwachsene?
Julian, 21, liebt die Musik und hat inzwischen seine eigene Band. Marieke, knappe drei, hat einen ausgeprägtem Willen und erforscht mit Begeisterung ihre Umgebung. Moritz Erichsen ist Ende dreißig und inzwischen Familienvater. Konstantin ist 13 und zählt in der Schule zu den Könnern am Kicker. Die vier kennen sich nicht, aber sie verbindet das gleiche Schicksal. Sie kamen mit einem komplexen Herzfehler zur Welt, einem Einkammerherzen. Um leben zu können, mussten alle mehrfach operiert werden.
"Ich weiß über meinen Herzfehler, als ich geboren bin, sah mein Körper so lila aus, so blaues und rotes Blut waren gemischt. Mit drei Operationen wurde das dann verändert. Das ist der Herzfehler, der die meisten Operationen, drei Operationen braucht. Aber ich merke sonst eigentlich keinen großen Unterschied zu anderen. Außer, dass ich jeden Abend Medikamente nehme und halt wöchentlich, oder wenn es mir nicht gut geht, so Blutentnahmen machen und die Blutgerinnung kontrollieren muss."
Franziska und Godeke Harrack haben zwei Töchter, Jannike und Marieke. Die kleine Marieke hat einen ähnlichen Herzfehler wie Konstantin, nur ist bei ihr nicht die linke, sondern die rechte Herzkammer betroffen. "In der zwanzigsten Schwangerschaftswoche war ich zum normalen Ultraschalltermin bei meiner Frauenärztin. Und die hat festgestellt, dass etwas mit dem Herzen nicht stimmt, konnte aber nicht genau sagen was und hat mich dann weiter zum Spezialisten geschickt."
Geheilt werden können besonders komplexe Herzfehler wie Einkammerherzen nicht. Damit Patienten trotzdem damit leben können, werden sie schon wenige Tage nach der Geburt zum ersten Mal operiert. Im Deutschen Herzzentrum München werden pro Jahr rund 25 bis 30 Kinder mit einem hypoplastischen Links- oder Rechtsherzsyndrom operiert. Bei drei notwendigen Operationen sind das bis zu 90 OPs, jede davon eine große Herausforderung für Herzchirurginnen wie Julie Cleuziou. "Die komplizierteste und die risikoreichste ist auf jeden Fall die erste Operation, weil im Neugeborenenalter stattfindet, wenn das Kind erst ein paar Tage alt ist. Das Gewebe ist da noch sehr zerreißlich. Das heißt, wir müssen noch vorsichtiger arbeiten als sonst. Es ist außerdem die größte Veränderung für den Kreislauf. Das ist ein Verfahren, das sehr komplex ist."
Bei einem hypoplastischen Herzsyndrom ist nur eine der beiden großen Herzkammern entwickelt. Venöses und arterielles Blut mischen sich, ein Überleben ohne OP ist nicht möglich. Beim hypoplastischen Links-Herzsyndrom ist die linke Kammer zu klein, beim Rechtsherzsyndrom die rechte. In den ersten zwei Lebensjahren wird in drei großen OPs das Herz regelrecht umgebaut. "Unser Ziel ist es, dass diese Kinder alle erwachsen werden und ein schönes Leben haben. Aber wir wissen natürlich, dass es nicht immer ein einfaches, unbedingt gutes Leben wird. Aber wir sehen immer wieder die Patienten, die kommen. Und wenn sie 20 sind und uns erzählen von ihrem Leben und man denkt, es ist so wie ein anderer junger Mensch, dann freuen wir uns sehr."
Julian ist heute 21 und lebt, wie seine jüngere Schwester, noch zu Hause bei seiner Mutter und deren Lebensgefährten. Auch er kam mit einem hypoplastischen Linksherzsyndrom zur Welt und wurde als Neugeborener und Kleinkind insgesamt dreimal operiert. "Ich habe immer gesagt, wenn Julian 21 wird, machen wir eine große Party. Einfach weil es ja immer kritische Momente gab, auch mal einen Moment, wo ich Freunde angerufen und gesagt habe, wir müssen uns von Julian verabschieden. Ich habe noch nicht ums Leben ringen müssen."
"Es gibt schon Situationen oder einfach Ängste generell, die man schon mit solchen Erfahrungen verbinden kann. Ja, das definitiv. "Musik begleitet mein Leben immer schon, in guten wie in schlechten Zeiten, Musik ist immer dabei. Und eigene Songs zu schreiben, ist eine coole Sache. Es bedeutet, sich ausdrücken zu können, sage ich jetzt mal. Alle Menschen mit komplexen Herzfehlern werden regelmäßig untersucht."
"Bei ihr sieht es gut aus. Ohne den eigenen Willen geht nichts. Und sie hat einen starken Willen. Das, was sie mitgemacht hat, war ja ziemlich schwierig, aber sie hat alles gepackt." Marieke wurde neun Tage nach ihrer Geburt zum ersten Mal operiert. Zuerst schien alles im Rahmen. "Dann kam es zu einer Situation, wo sie reanimationspflichtig wurde, weil die Sättigung so abgefallen war. Und dann wurde sie reanimiert, was auch nicht erfolgreich war. Und dann wurde die ECMO eingebaut. Also die Ärzte haben dann wirklich alles getan, um ihr Leben zu retten und drei Tage später konnte die ECMO auch wieder ausgebaut werden. Und dann hat sie sich Schritt für Schritt wieder zurückgekämpft. Denn auch wenn alles glatt läuft, Garantien gibt es in der Medizin nicht, meint Prof. Julie Cleuziou, es könne immer etwas passieren. Patienten mit komplexen Herzfehlern haben statistisch gesehen geringere Überlebenschancen als Patienten mit weniger komplexen, einfacheren Herzfehlern.
Moritz Erichsen ist 38 Jahre alt, verheiratet und hat zwei Kinder. Auch er kam mit einem hypoplastischen Rechtsherzsyndrom auf die Welt. Er ist einer der Pioniere mit diesem komplexen Herzfehler. Als er Mitte der 80-ger Jahre geboren wurde, hatten Kinder wie er in Deutschland so gut wie keine Überlebenschance. Dass er lebt, verdankt er dem Einsatz seiner Eltern. Nach seiner Geburt kam er als Notfall per Hubschrauber nach München. Doch die Ärzte konnten nicht helfen. Es sei nur möglich, sein Leben etwas zu verlängern, so ihre einhellige Einschätzung. "Aber Moritz, der blieb und blieb und blieb, während andere leider auch verstarben. Er hat einfach den Lebenswillen gehabt. Seine Eltern entschließen sich zur OP. Das Problem: Die Eingriffe sichern sein Überleben maximal für die ersten zwei, drei Jahre, mehr war damals in Europa nicht möglich. Durch Zufall bekommen sie einen Hinweis, dass es in einer Klinik in Boston einen Kinderherzchirurgen, Professor Castaneda gäbe, der große Erfahrung mit OPs von Herzfehlern habe. Sie schreiben ihm und schicken, was sie an Unterlagen haben. Vierzehn Tage später kommt tatsächlich ein Telegramm. Sie sollen mit ihrem Sohn in die USA kommen. Dort könne er operiert werden. Allerdings ist die Methode neu, riskant und gefährlich. Die Überlebenschancen für Moritz Erichsen liegen bei 40 zu 60 Prozent. Die Operation gelingt, Moritz Erichsen überlebt. "Ich bin einfach megastolz auf meine Eltern! Und für das, was sozusagen passiert ist, für das, was ich hatte und habe, führe ich ein gutes Leben. Es ist zwar kein ganz normales Leben, das muss man ehrlicherweise sagen. Als Kind geht man da relativ normal mit um, also als Kleinkind oder kleines Schulkind. Je älter man dann wird, merkt man dann schon so ein bisschen auch seine Limitation, die man hat im Vergleich zur gleichen Altersgruppe."
Heute ist die Methode weltweit etabliert. Moritz Erichsen ist Patient der Klinik für angeborene Herzfehler des Deutschen Herzzentrums München. Inzwischen weiß man: Menschen mit angeborenen Herzfehlern sind chronisch krank, egal ob die Fehler korrigiert, ob sie leicht oder hochkomplex sind. Für Menschen wie Moritz Erichsen ist besonders wichtig, von Ärzten behandelt zu werden, die Spezialisten für Menschen mit angeborenen Herzfehlern sind. In großen Zentren wie dem Deutschen Herzzentrum München liegen deshalb Kleinkinder, Kinder, Erwachsene und auch ältere Menschen mit angeborenen Herzfehlern mitunter auf ein und derselben Station. "Es für uns natürlich eine erfreuliche Entwicklung, dass diese Patienten inzwischen gut behandelt werden können."
Denn es gibt gesundheitliche Risiken und Langzeitfolgen, deren Behandlung auch erfahrene Mediziner vor große Herausforderung stellt. Prof. Peter Ewert, Direktor der Klinik für angeborene Herzfehler des Deutschen Herzzentrums München sieht die positive Entwicklung als großes Experiment mit bisher erfreulichem Verlauf. Moritz Erichsen sind die gesundheitlichen Risiken seiner Erkrankung bewusst, er ist heute selbst Arzt. "Es gibt natürlich immer die Risiken Rhythmusstörungen und solche Sachen, wie zum Beispiel Probleme mit der Leber bis hin zu langfristigen Leberveränderungen. Klar, über so was macht man sich dann schon auch immer mehr Gedanken. Meine Familie bedeutet natürlich alles für mich. Das ist dann nochmal eine ganz andere Dimension. Wenn man Kinder hat, da fragt man sich natürlich, wie die Zukunft sein wird. Aber letzten Endes wird das mittlerweile auch alles erst herausgefunden. Ich bin eben einer der Patienten, die jetzt so lange schon leben."
Aufgewachsen ist Moritz Erischsen als zweites von vier Geschwistern, ganz normal. Lassen sie ihn machen, was er will, hatte sein behandelnder Kardiologe seinen Eltern geraten. Wozu hätte er denn sonst alle Operationen und Behandlungen durchgemacht? Ein Rat, an den sich Ruth und Axel Erichsen trotz manch riskanter Unternehmungen ihres Sohnes über die Jahre gehalten haben. Und darauf sind sie mit Recht im Rückblick stolz.
Das inzwischen gewonnene, medizinische Wissen kommt Familien von heute zugute. Vor wenigen Monaten hatte Marieke ihre letzte Operation. Wie viele Familien wurde und wird Familie Harrack auch nach dem letzten Klinikaufenthalt durch das Zentrum univentrikuläres Herz und andere komplexe Herzfehler des Deutschen Herzzentrums München zu Hause weiterbetreut. "Die Familien lernen unterschiedlich schnell, mit den unterschiedlichen Herausforderungen zu leben. Wir sind auch nach dem Aufenthalt in der Klinik immer wieder im Kontakt. Gerade in der Winterzeit haben viele Kinder Fieber. "Es sind weit über 300 Familien, die wir begleitet haben."
Angeborene Herzfehler - Wie man sie erkennt und behandelt | Health Celerates
Pränatale Diagnose und Beratung
Dr. med. Tiffany Kogler ist selbst promovierte Ärztin und erwartet ihr zweites Kind. Alles sei in bester Ordnung, hatte ihre Frauenärztin gemeint. Doch sie bestand trotzdem auf einer weiteren Ultraschall-Untersuchung und einer zweiten Meinung. "Es war die ganze Schwangerschaft schon immer so ein Gefühl, dass ich gesagt habe, irgendetwas ist nicht wie beim ersten Mal. Und dann kommt der Arzt, schaut im Ultraschall und sagt, da haben wir ein Problem. Es ist ein großer Schock." Ihr zweites Kind hat ein hypoplastisches Linksherzsyndrom. Das kann mit einem entsprechend hochauflösendem Ultraschall schon ab der zwölften Schwangerschaftswoche erkannt werden; vorausgesetzt, der oder die Untersuchende kennt sich mit angeborenen Herzfehlern aus.
Bestätigt sich die Diagnose eines Herzfehlers, werden Eltern von Expertinnen wie Prof. Renate Oberhoffer-Fritz von der Präventiven Pädiatrie der TU München untersucht und beraten. Die gute Nachricht dabei ist: Auch Kinder mit einem solch komplexen Herzfehler wie einem hypoplastischen Herzsyndrom entwickeln sich im Mutterleib vollkommen normal. "Solange das Kind im Mutterleib ist, geht es ihm hervorragend, weil es diverse Querverbindungen des rechten und linken Kreislaufs zueinander gibt, die das Überleben ermöglichen."
Für Ärztinnen wie Prof. Renate Oberhoffer-Fritz, die Familien in der schwierigen Zeit zwischen Diagnose und der Geburt begleitet, ist die Aufklärung der werdenden Eltern keine leichte Aufgabe. Denn die Perspektiven nach der Geburt sind oft ungewiss. Manche Frauen und Familien entscheiden sich für einen Schwangerschaftabbruch. "Wir können leider nicht im Detail vorhersagen, welches Kind wird ein Problem bekommen und welches entwickelt sich optimal. Ich muss versuchen, die ganze Bandbreite des Möglichen aufzuklären und darzustellen, damit die Familien allumfassend wissen, was auf sie zukommen kann. Und damit sie sich ein Bild machen können, was sie schaffen, was sie tragen können und wollen. Manche Familien sagen, das schaffen wir nicht. Dann muss ich die andere Richtung auch entsprechend darstellen. Da muss die psychosoziale Beratung hinzu. Also der psychologische Zustand der Mutter, das ist ja der Hauptgrund für den Schwangerschaftsabbruch, nicht die Fehlbildung an sich. Da hat sich das Recht in Deutschland geändert. Und es muss natürlich Kollegen geben, die so einen Abbruch dann auch machen. Das ist dann auch abhängig vom Zeitpunkt. Kommen die Frauen wirklich in dem Moment, wo das Kind, würde es zur Welt kommen, nicht mehr lebensfähig ist, oder ist die Grenze schon überschritten? Das ist manchmal eine sehr schwierige und sehr diffizile Situation. Aber alle Familien brauchen Hilfe."
Dr. Zwar weiß man heute, dass genetische Faktoren eine Rolle für die Entstehung komplexer angeborener Herzfehler spielen, aber wann und warum es zu Fehlentwicklungen kommt, ist wissenschaftlich noch nicht geklärt. Für die Familien spielt das keine große Rolle, denn sie leben mit der Erkrankung ihren Alltag. Konkrete Informationen und andere wertvolle Angebote bieten Selbsthilfegruppen wie der Verein Junge Herzen Bayern. Eltern und Kinder nutzen gerne die Möglichkeit, sich auf Freizeiten mit anderen auszutauschen, die ähnliche Herausforderungen meistern.
Moritz Erichsen ist zuversichtlich, dass sein gesundheitlich stabiler Zustand noch viele Jahre anhält. Konstantin sagt, er führt ein Leben, das sich nicht wesentlich von dem seiner Altersgenossen unterscheidet. Er spielt nicht nur Tischtennis. Auch in 20 Aufführungen der Passionsspiele Waal stand er mit auf der Bühne. Die Eingewöhnungsphase verlief ohne Probleme und auch ihre Mutter freut sich darüber, wieder ihrem Beruf nachgehen zu können. Und Julian hat inzwischen eine Ausbildung als Erzieher begonnen. "Es wäre schon ziemlich schade, wenn ich nicht da wäre!"